
Short Biography
Winship Herr received his PhD from Harvard University in 1982 for studies on recombinant retroviruses in leukemogenic mice with Walter Gilbert. After postdoctoral studies with Frederick Sanger in Cambridge, UK and Joe Sambrook at Cold Spring Harbor Laboratory, he joined the Cold Spring Harbor Laboratory faculty in 1984. There he served as assistant director of the Laboratory from 1994-2002 and from 1998-2004 was the founding dean of the Watson School of Biological Sciences, a doctoral degree-granting school. He arrived at the CIG in 2004. Within the UNIL Faculty of Biology and Medicine, he was vice-dean for Biology (2007-2009) and director of the School of Biology (2009-2015). He was elected member of the European Molecular Biology Organization (EMBO) in 2008.

WINSHIP HERR – RESEARCH REPORT
Mammalian transcriptional regulation
Two complete sets of instructions contained within the genomes we inherit from our parents are responsible for directing a single cell — the zygote — to become an adult human being. This process results from controlled patterns of gene expression that are maintained, as well as changed, during many rounds of cell division, differentiation, and death. Control of gene transcription is fundamental to these processes, with genetic and epigenetic defects in transcriptional regulation often leading to human disease, including cancer.
HCF-1
To investigate these processes, we study a key regulator of human cell proliferation, called HCF-1 for herpes simplex virus host-cell factor-1. It serves as an adaptor protein that links a large number of site-specific DNA-binding proteins with a plethora of chromatin-modifying activities, resulting in both activation and repression of transcription. It also undergoes an unusual proteolytic maturation process that generates two associated HCF-1 subunits that regulate different phases of the human cell cycle: An N-terminal subunit permits cells to progress into S phase for genome replication and a C-terminal subunit promotes proper segregation of the replicated genome in M phase.
O-GlcNAc transferase possesses separable mechanisms for glycosylation and HCF-1 proteolysis
HCF-1 is cleaved at a critical glutamate residue located within any one of six 26 amino acid repeated sequences, called HCF-1PRO repeats. We discovered previously that the enzyme responsible for HCF-1 cleavage is O-linked β-N-acetylglucosamine transferase (OGT), an enzyme previously known only for modification of a large number of cytoplasmic and nuclear proteins by the addition of the sugar N-acetylglucosamine (GlcNAc) to serine or threonine residues in the form of O-GlcNAc. OGT not only cleaves but also heavily glycosylates HCF-1. Thus, a single enzyme can both cleave and chemically modify a protein substrate.
In 2013, in collaboration with Dr. S. Walker of Harvard University Medical School, we showed that OGT-induced glycosylation and HCF-1 proteolysis occur in the same active site of OGT. Indeed, the HCF-1PRO-repeat glutamate residue targeted for cleavage resides in the same position within the catalytic site as a targeted serine of a glycosylation substrate, such that conversion of the HCF-1 cleavage-site glutamate into serine converts the HCF-1PRO repeat into a glycosylation substrate. This shows how intimate the boundary between reversible (i.e., glycosylation) and irreversible (i.e., proteolysis) post-translational modification can be.
Whereas the aforementioned findings identified many similarities between OGT glycosylation and HCF-1 cleavage, we have now shown that there are significant differences. For example, we showed that mutations within OGT could inactivate proteolysis without evident effects on glycosylation (Figure 1) and vice versa. Indeed, in collaboration with Dr. Daan van Aalten of the University of Dundee, we showed that even the OGT co-substrate for glycosylation, UDP-GlcNAc — while essential for proteolysis — plays roles in glycosylation not required for proteolysis.
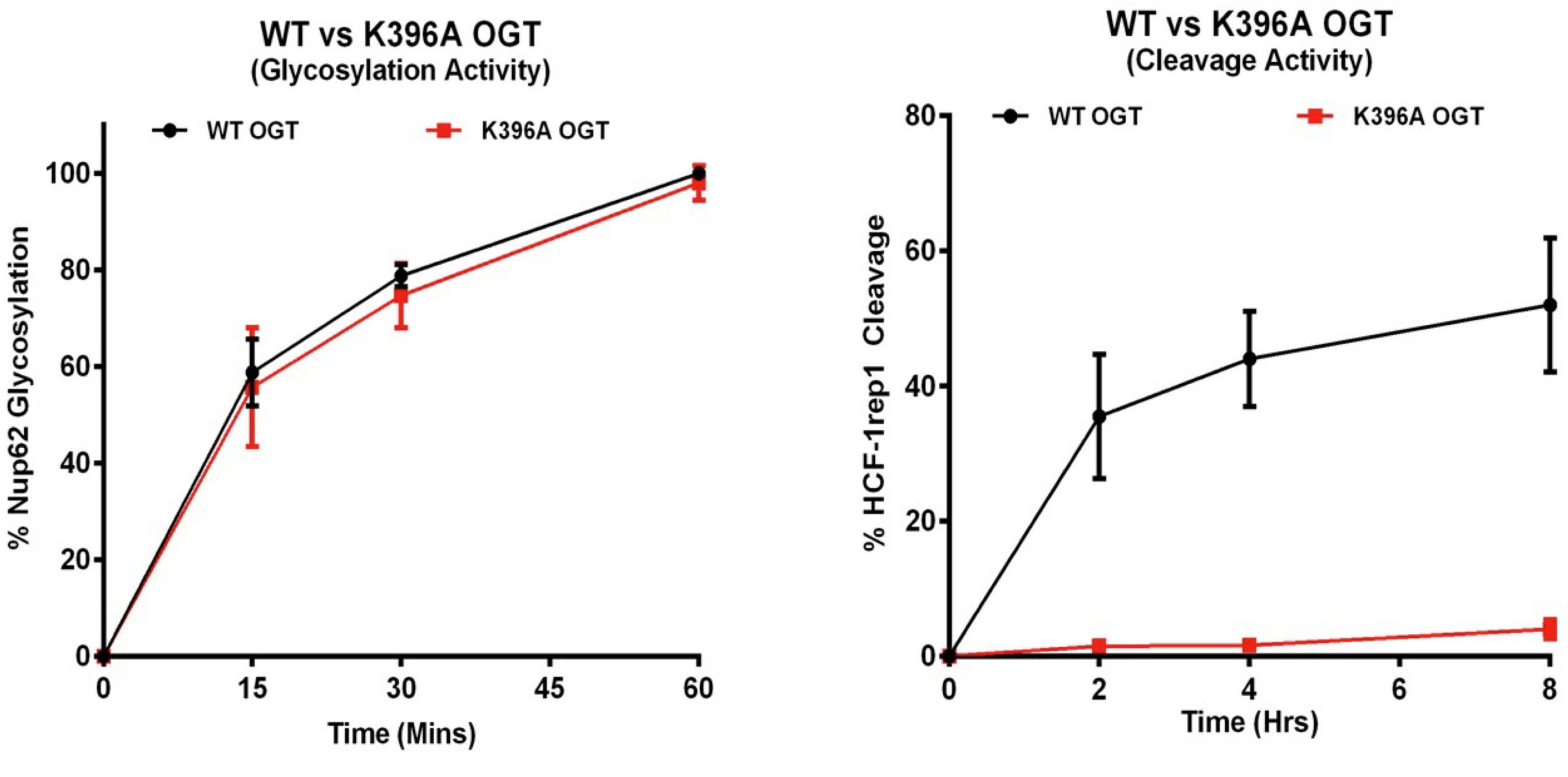
Fig 1. A single point mutation, K396A, in OGT disrupts HCF-1PRO-repeat-cleavage (right) but not glycosylation (left). (See text and Kapuria et al. (2016) Genes Dev. 30, 960–972 for details.)
We also discovered that even OGT’s from invertebrates — where the HCF-1 homolog is not subject to OGT-mediated proteolysis — can cleave human HCF-1, indicating that the HCF-1PRO repeat targets conserved elements of OGT for its proteolysis. These results have led us to propose that the HCF-1PRO repeat evolved to be cleaved by OGT as opposed to OGT having evolved to cleave HCF-1.
Compensatory embryonic response to allele specific inactivation of the murine X-linked gene HCFC1
Early in female mammalian embryonic development cells randomly inactivate gene expression from one of the two X chromosomes, to compensate for the presence of two X chromosomes as opposed to the single X chromosome found in males. The mouse gene encoding HCF-1, called Hcfc1, is X-linked. By generating a Cre-recombinase inducible Hcfc1 knock-out (KO) allele, we have probed the role of HCF-1 in actively proliferating embryonic cells and for cell-cycle re-entry during liver regeneration. In resting adult liver cells, the requirement for HCF-1 in cell proliferation is evident as hepatocytes lacking HCF-1 fail to re-enter the cell cycle and thus to proliferate during liver regeneration (Figure 2). HCF-1 function is also essential for embryonic development. Nevertheless, in heterozygous wild-type and KO female embryos, Hcfc1 deletion around embryonic day 5.5 (E5.5) is well tolerated; it leads to a mixture of HCF-1-positive and -negative embryonic cells owing to random X-chromosome inactivation of the wild-type or KO Hcfc1 allele. In subsequent days of development, however, HCF-1-negative cells disappear owing to cell-cycle exit and apoptosis. Although generating a temporary developmental retardation, the loss of HCF-1-negative cells is tolerated, leading to viable heterozygous offspring with skewed inactivation of the X-linked Hcfc1 KO allele. The survival of the heterozygous female embryos, even with half the cells genetically compromised, illustrates the developmental plasticity of the post-implantation mouse embryo — in this instance, permitting survival of females heterozygous for an X-linked embryonic lethal allele.
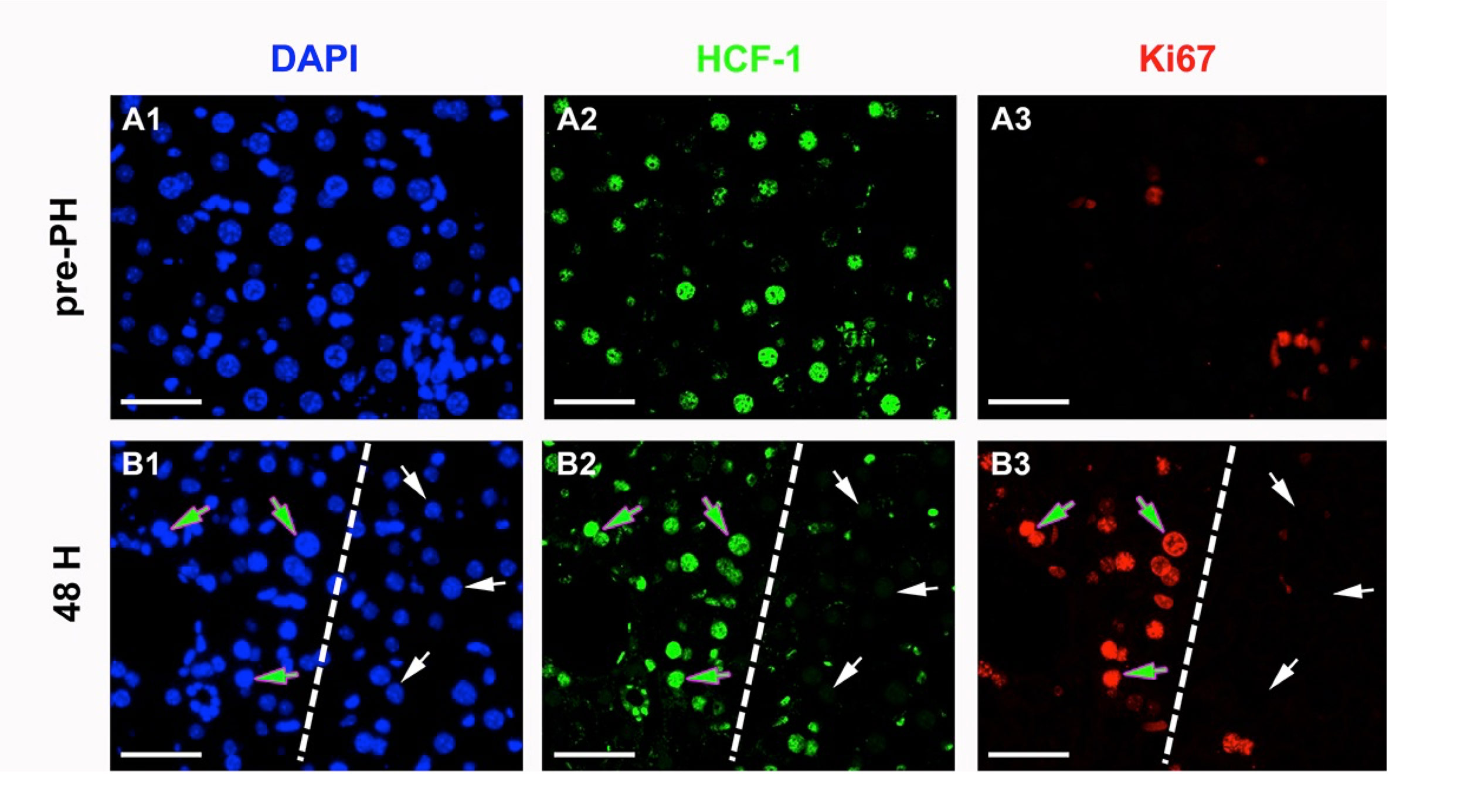
Fig 2. Hepatocytes lacking HCF-1 fail cell-division cycle entry upon partial hepatectomy. Immunofluorescence for cell-cycle progression markers in heterozygous Hcfc1 wild type and knock out female mice before (panels A) and 48 h after (panels B) being subjected to 70% partial hepatectomy. The paraffin-embedded liver sections were treated with DAPI for DNA (blue), anti-HCF-1 antibodies (green), and the Ki67 cell-proliferation marker (red). The broken lines in panels B1–3 separate the HCF-1-positive from HCF-1-negative hepatocytes. The green arrows point to DAPI-, HCF-1- and cell-proliferation marker-positive hepatocytes, and the white arrows point to DAPI-positive, HCF-1-negative and cell-proliferation marker-negative hepatocytes. Scale bar: 50 μm.
Epiblast-specific loss of HCF-1 leads to failure in anterior-posterior axis specification
In collaboration with Dr. Daniel Constam at the Ecole Polytechnique Fédérale de Lausanne, we showed that — whereas E5.5 heterozygous Hcfc1 knock-out female embryos can survive, owing to the replacement of all HCF-1-deficient cells by HCF-1-positive cells during following development — complete E5.5 HCF-1 loss in hemizygous male embryos leads to embryonic arrest at E6.5 with a rapid progressive cell-cycle exit and an associated failure of anterior visceral endoderm migration and primitive streak formation. Subsequently, gastrulation does not take place. We noted that the pattern of embryonic lethality displays many similarities to loss of b-catenin function, suggesting a relationship between HCF-1 function and Wnt signaling.
GROUP MEMBERS
Group Leader
Winship Herr
winship.herr@unil.ch
Bioinformatician
Viviane Praz
Postdoctoral Fellows
Daria Gudkova
Vaibhav Kapuria
Shilpi Minocha
Maria Leonor Rib Perez
PhD Students
Harmonie Dehaene
Laura Sposito
Technicians
Maykel Duarte Lopes
Fabienne Lammers
Philippe Lhote
Danièle Pinatel
Dominic Villeneuve
Visiting Student
Annabelle Brodeur
Summer Student
Joseph Samson Bowness
Administrative assistant Nathalie Clerc
nathalie.clerc@unil.ch