
Short Biography
Alexandre Reymond carried out his thesis in the laboratory of Dr. Viesturs Simanis at the Swiss Institute for Experimental Cancer Research (ISREC) and received his PhD from the University of Lausanne in 1993. After completion of his postdoctoral training with Dr Roger Brent in the Department of Molecular Biology, Massachusetts General Hospital and in the Department of Genetics, Harvard Medical School in Boston, he moved to the Telethon Institute of Genetics and Medicine (TIGEM) in Milan in 1998 to lead a research group. He joined in 2000 the Department of Genetic Medicine and Development, University of Geneva Medical School. He moved to the Center for Integrative Genomics in October 2004 and became its Director in 2015.

ALEXANDRE REYMOND – RESEARCH REPORT
Assessing the impact of copy number variants
The completion of the human genome sequence and technological advances allowed pinpointing that any two human individuals differ on approximately 0.8% of their genome length. With such extent of genomic sequences concerned, DNA copy number variants (CNVs), i.e. stretches of DNA that display altered genomic copy numbers, significantly contribute to phenotypic variation and disease predisposition. Large recurrent CNVs with rare population prevalence were found to be associated with genetic disorders, particularly neurodevelopmental traits such as intellectual disability. For example, 69 distinctive recurrent structural rearrangements, often regrouped under the term “genomic disorder” coined by Jim Lupski, are listed by the DECIPHER Database (http://decipher.sanger.ac.uk/).
Global influence of structural variation
The association of CNVs with disorders such as intellectual disability or ASD (autism spectrum disorder) has been almost exclusively evaluated using clinically ascertained cohorts. Thus, the contribution of these genetic variants to cognitive phenotypes in the general population remains unclear. To investigate the clinical features conferred by CNVs associated with known syndromes in adult carriers without clinical pre-selection, we analyzed the Estonian Genome Center’s cohort that comprises 5% of the Estonian adults and is representative of the country’s population. We observe that the phenotypes of carriers of known genomic disorder lesions, such as the 16p11.2 CNVs, are reminiscent of those of carriers of identical rearrangements identified in disease cohorts, challenging the opinion that carriers identified in control cohorts present normal or sub-clinical phenotypes. We also assessed the genome-wide consequences of rare intermediate size CNVs (frequency ≤0.05%; size ≥250kb). We found associations between rare CNVs and lower educational attainment and intellectual disability prevalence in the general population. These results were replicated by analyses of cohorts of adults from Italy and the United States and adolescents from the United Kingdom. These results were published in JAMA (Männik et al., 2015).
A potential contributory role for ciliary dysfunction in the 16p11.2 600kb BP4-BP5 pathology
The 16p11.2 600kb BP4-BP5 copy number variants (CNVs) are associated with mirror phenotypes on BMI, head circumference and brain volume and represent frequent genetic lesions in ASD and schizophrenia. We interrogated the transcriptome of individuals carrying reciprocal 16p11.2 600kb BP4-BP5 CNVs. Transcript perturbations correlated with clinical endophenotypes and were enriched for genes associated with ASD, abnormalities of head size and ciliopathies. Ciliary genes expression was also perturbed in orthologous mouse models, raising the possibility that ciliary dysfunction contributes to 16p11.2 600kb BP4-BP5 pathologies. In support of this hypothesis, we found structural ciliary defects in the CA1 hippocampal region of 16p11.2 600kb BP4-BP5 duplication mice. Moreover, using an established zebrafish model we show genetic interaction between KCTD13, a key driver of the mirrored neuroanatomical phenotypes of the 16p11.2 600kb BP4-BP5 CNV and ciliopathy-associated genes. Overexpression of BBS7 rescues head size and neuroanatomical defects of kctd13 morphants, while suppression or overexpression of CEP290 rescues phenotypes induced by KCTD13 under- or overexpression, respectively. Our data suggest that dysregulation of ciliopathy genes contributes to the clinical phenotypes of these CNVs. These results were published in the Am J Hum Genet (Migliavacca et al., 2015).
Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes
Copy number variants (CNVs) are major contributors to genomic imbalances disorders. Phenotyping of 137 unrelated deletion and reciprocal duplication carriers of the distal 16p11.2 220kb BP2-BP3 interval showed that these rearrangements are associated with autism spectrum disorders (ASD) and mirror phenotypes of obesity/underweight and macro-/microcephaly. Such phenotypes were previously associated with rearrangements of the non-overlapping proximal 16p11.2 600kb BP4-BP5 interval. These two CNVs-prone regions at 16p11.2 are reciprocally engaged in complex chromatin looping, as successfully confirmed by 4C-seq, FISH and Hi-C, as well as coordinated expression and regulation of encompassed genes. We observed that genes differentially expressed in 16p11.2 BP4-BP5 CNV carriers are concomitantly modified in their chromatin interactions, suggesting that disruption of chromatin interplays could participate in the observed phenotypes.
We also identified cis- and trans-acting chromatin contacts to other genomic regions previously associated with analogous phenotypes. For example, we uncovered that individuals with reciprocal rearrangements of the trans-contacted 2p15 locus similarly display mirror phenotypes on head circumference and weight. Our results indicate that chromosomal contacts’ maps could uncover functionally and clinically related genes. These results were published in Molecular Psychiatry (Loviglio et al., 2016).
Homo sapiens-specific gene family expansion and the evolution of autism risk at chromosome 16p11.2
Recurrent pathogenic deletions and duplications at chromosome 16p11.2 are mediated by a complex set of highly identical and directly oriented segmental duplications. This disease-predisposing architecture results from recent, Homo sapiens-specific duplications (i.e. absent in Neandertal and Denisova) of a segment including the BOLA2 gene, the latest among a series of genomic changes that dramatically restructured the region during hominid evolution. All humans ( n = 2,359) examined carry one or more copies of the duplication, which nearly fixed early in the human lineage — a pattern unlikely to have arisen so rapidly in the absence of selection (p < 0.0097). BOLA2 copy number correlates with both RNA expression (r = 0.36) and protein level (r = 0.65), with the greatest expression difference between human and chimpanzee in experimentally derived stem cells. Analyses of 152 patients carrying a 16p11.2 rearrangement showed that >96% of breakpoints occur within the Homo sapiens-specific duplication. In summary, the duplicative transposition of BOLA2 at the root of the Homo sapiens lineage ~282 kya simultaneously increased copy number of a gene associated with iron homeostasis and predisposed our species to recurrent rearrangements associated with disease. These results that stem from a fruitful collaboration between Evan Eichler’s and my lab were recently published in Nature (Nuttle et al., 2016).
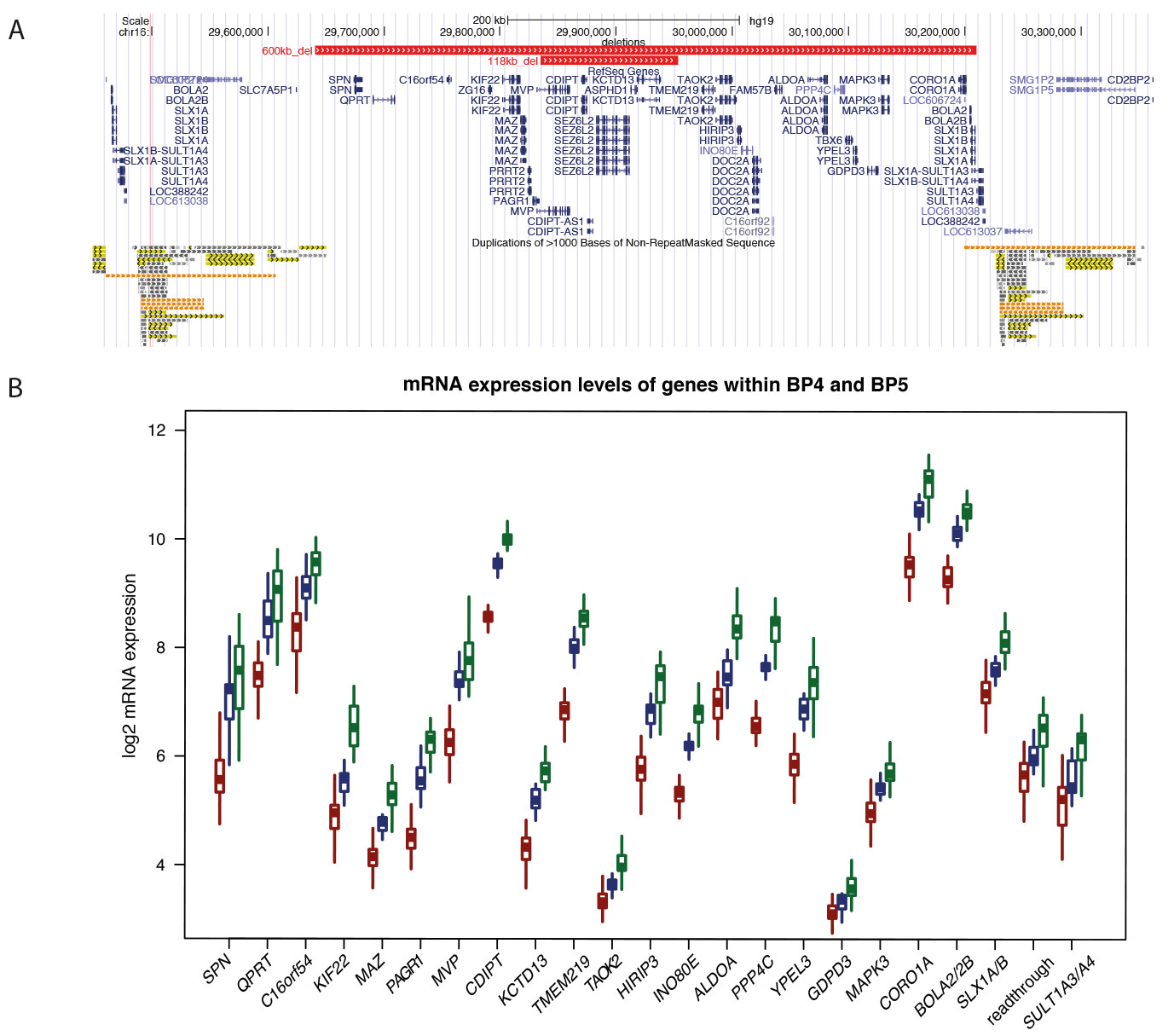
Fig 1. Expression levels changes in 16p11.2 deletions and duplications LCLs. (Migliavacca et al., 2015)
(A) Comparison of the GRCh37/hg19 genomic boundaries of the recurrent 600kb BP4-BP5 16p11.2 deletion with that of the 118kb atypical deletion described in 15. The genes mapping within the interval are shown in blue. The low-copy repeats (LCR) at the rearrangements breakpoints are indicated at the bottom.
(B) Relative expression levels measured by microarrays in deletion (n=50) and duplication (n=31) carriers (red and green, respectively), and control LCLs (n = 17, blue) of the 19 “unique” and 3 multiple-copy genes (plus the read-through transcription between SLX1A/1B (MIM #615822/615823) and SULT1A3/A4 (MIM #600641/615819)) expressed in LCLs and mapping within the 16p11.2 BP4-BP5 interval and its flanking LCRs, respectively. Note that the expression level of these transcripts, de facto positive controls, is positively correlated with gene dosage.
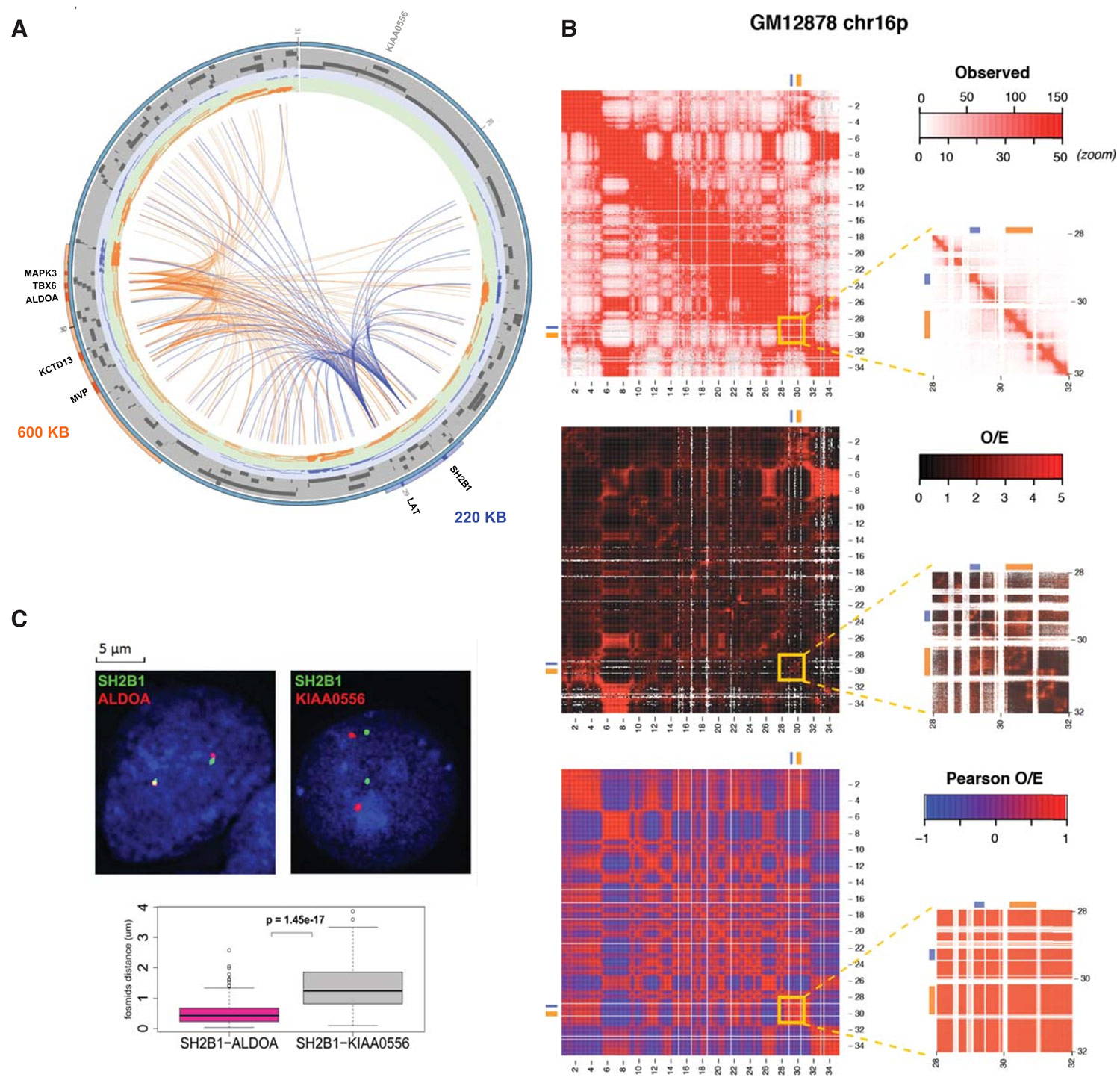
Fig 2. Chromatin interactions between the 16p11.2 600kb BP4-BP5 and 220kb BP2-BP3 genomic intervals. (Loviglio et al., 2016)
(A) Circos plot representation of the chromatin loops identified in the human chromosome 16 27.5 to 31.0Mb window. The 220kb BP2-BP3 and 600kb BP4-BP5 intervals are depicted by blue and orange bars on the peripheral circle, respectively. Darker sections indicated the positions of the viewpoints. Central blue and orange lines indicate the chromatin interactions corresponding to BP2-BP3 and BP4-BP5 viewpoints, respectively. Note the quasi absence of loops between the BP2-BP3 viewpoints (LAT and SH2B1) and the 27.5-28.4Mb region. The mapping position of the KIAA0556 gene, used as control locus in FISH experiments, is indicated.
(B) High resolution Hi-C chromosome conformation capture results obtained in reference 75 with the GM12878 LCL within the chromosome 16 0-34Mb window (left panels) and zoom in within the 28-31 Mb region encompassing the two CNVs (5kb resolution; right panels). The positions of the 220kb BP2-BP3 and 600kb BP4-BP5 intervals are shown by blue and orange bars, respectively. Observed (top panel), observed/expected (central panel) and Pearson correlation results are presented (bottom panel).
(C) Fluorescence in situ hybridization experiments show colocalization of SH2B1 foci (green) that map to the 220kb BP2-BP3 interval with ALDOA foci (red) that map to the 600kb BP4-BP5 genomic interval (left panel) but not with the equidistant KIAA0556 (red) foci (central panel). The distribution of interphase nuclei distances between the SH2B1 and ALDOA (deep pink) and SH2B1 and KIAA0556 foci (grey) are shown in the lower panel. The mapping positions of ALDOA, SH2B1 and KIAA0556 are indicated in (A).
GROUP MEMBERS
Group Leader
Alexandre Reymond
alexandre.reymond@unil.ch
SNSF Ambizione Fellow
Binnaz Yalcin
SNSF Marie Heim-Vögtlin Fellow
Catia Attanasio
Postdoctoral Fellows Giuliana Giannuzzi
Lucie Gueneau
Katrin Männik
Konstantin Popadin
Eleonora Porcu
PhD Students
Pasquelena De Nittis
Anne-Maud Ferreira
Andreas Gschwind
Anna Mikhaleva
Nicla Loviglio
Namrata Sarkar
Norine Voisin
Marianna Zazhytska
Master Student
Ilse van der Werf
Technicians
Jacqueline Chrast
Michaël Wiederkehr
Administrative Assistants
Annick Crevoisier
Iris Marouani
iris.marouani@unil.ch