
Short Biography
Paul Franken received his PhD in 1993 for his work on sleep homeostasis and thermoregulation at the University of Zürich, under the direction of Alexander Borbély. He then joined the group of Craig Heller at Stanford University as postdoctoral fellow, where he studied the cellular mechanisms underlying circadian clock resetting. In 1996, he joined Mehdi Tafti at the University of Geneva where he used QTL analysis to map sleep and EEG traits in mice. He then moved back to Stanford in 2000 as a senior research scientist to establish an independent lab. He continued to work on the genetics of sleep homeostasis and further focused on the molecular interactions between circadian rhythms, sleep homeostasis and brain metabolism. In 2005, he joined the CIG as Maître d’enseignement et de recherche and was promoted Associate Professor in 2013. Current focus of the Franken lab is on the role of sleep-wake states in driving clock gene expression and on systems genetics approaches to dissect the molecular pathways critical in the response to acute sleep loss.

PAUL FRANKEN – RESEARCH REPORT
Genetics and energetics of sleep homeostasis and circadian rhythms
Two main regulatory processes have to be considered when studying sleep and its disorders: a sleep-wake driven homeostatic process that is activated by and counters the effects of sleep loss, and a circadian process that determines the time-of-day sleep occurs. The fine-tuned interaction between the two permits us to stay awake and alert throughout the day and to remain asleep at night. Relative minor changes in this interaction can already lead to performance decrements and clinically significant sleep disruption. To gain insight in the molecular pathways underlying the sleep homeostat and its interaction with the circadian process, we apply forward, molecular, and reverse genetic approaches in the mouse, using sleep deprivation (SDep) as a tool to challenge the sleep homeostat.
Clock genes signal both time-of-day and time-spent-awake
We discovered that the handful of genes known to set circadian time (aka as clock genes) are also involved in the homeostatic regulation of sleep. Thus, in mice lacking one or more of the core clock genes (e.g. Npas2, Bmal1, Cry1, and Cry2), the sleep homeostatic response to SDep is altered. Moreover, changes in the forebrain expression of the clock genes Per(iod)1 and -2 are tightly linked to the prior sleep-wake history. We demonstrated that these changes in Per1 and -2 expression are, in part, mediated by their transcriptional regulator NPAS2 and that SDep reduces the specific DNA binding of NPAS2 to the E-boxes of its target genes such as Per1 and -2. Thus contrary to the prevailing notion that circadian and homeostatic processes are separate, at the cellular level the same molecular circuitry seems to be implicated in both. We now focus on the mechanisms linking clock gene expression to time-spent-awake (and -asleep). Because the transcriptional activity of e.g. NPAS2 depends on intracellular energy charge, the molecular circadian clock could directly link cellular metabolism to homeostatic sleep need. We have investigated this issue in vitro using redox-sensitive GFP probes and in vivo by simultaneously monitoring PER2 protein, locomotor activity, and EEG in freely behaving mice. Moreover, we discovered that the stress hormone corticosterone greatly contributes to the transcriptome changes in the brain after SDep and that of Period gene expression in particular. Using mathematical modeling we could reliably quantify the complex relationship between changes in clock gene expression, the sleep-wake distribution, and circulating corticosterone levels (Figure 1). Model predictions are useful in helping to design relevant experiments to unravel these non-linear relationships.
Micro-RNAs and sleep homeostasis
Micro-RNAs (miRNAs) have received much interest in neuroscience. Given that these non-coding transcripts importantly affect the mRNA levels of many coding genes, we investigated the role of miRNAs in sleep homeostasis using two approaches. First, we made an inventory of miRNAs affected by SDep in various brain areas and identified miR-709 to be strongly and consistently up-regulated after SDep. Inhibiting miR-709 resulted in altered electroencephalogram (EEG) delta (1-4 Hz) activity after SDep, suggesting an altered sleep homeostat. In a second approach, we assessed sleep homeostasis in mice lacking Dicer, which encodes an enzyme critical for miRNA maturation. We used a conditional Dicer knockout construct with which miRNAs can be down-regulated specifically in neurons of adult mice. Besides a profound homeostatic phenotype these mice also displayed an unexpected obesity phenotype (Figure 2). Thus in addition to their now established role in circadian rhythm generation, we show that miRNAs also play a role in the homeostatic aspect of sleep regulation.
Systems genetics of acute sleep loss
We have previously shown that genetic factors importantly shape sleep and brain activity (EEG). Quantitative trait loci (QTL) mapping in inbred mice led to the identification of the responsible genes underlying three EEG-related traits, Acads, Rarb, and Homer1a, thereby implicating novel signaling pathways involved in rhythmic brain activity and sleep need. We continued this line of research with two large scale projects.
* In a first, recently completed project, we used the genetically diverse (outbred) CFW mice to map genes implicated in sleep with gene-level resolution. Using a high-throughput sleep phenotyping technique we helped develop, we measured sleep in 1613 mice and obtained strong, genome-wide significant signals for sleep quality, directly implicating Unc13c, which is involved in synaptic transmission, and Pgc1α, which is associated with cortical hyper-excitability.
* In a second, ongoing project, we took advantage of a genetic reference population, the BXD panel of recombinant inbred lines, for a systems genetics approach. We have interrogated 33 lines of this panel at the genome, chromatin, transcriptome, metabolome, behavioral, EEG, tissue, and cellular levels to chart the core genetic networks important to sleep and its homeostatic regulation. A short inventory of the results demonstrates both the pervasive effects of sleep loss and of genetic factors: 61 phenotypic phQTLs, 22 metabolic mQTLs, and ca. 10’000 cis–eQTLs were identified and the short (6h) sleep deprivation we performed significantly changed the levels of 51% of the metabolites quantified and up to 78% of all expressed transcripts! Also this differential gene expression was under strong genetic control with ca. 110 cis– and 2000 trans–eQTLs. The integrative, systems genetics aspect of this project can be illustrated with one phenotypic trait; the increase in NREM sleep time after sleep deprivation (Figure 3). This trait greatly varied among the BXD lines yielding a significant QTL on Chr 4. Unbiased prioritization scoring identified Acot11 as top candidate. Moreover, a cis-eQTL affecting Acot11 expression after sleep deprivation in liver specifically, was found and the responsible SNP predicted the increase in NREM sleep after sleep deprivation. Strain variation in the fatty acid phosphatidylcholine-ae-C38:2 also mapped to the same genomic region and its plasma levels after SDep strongly correlated with Acot11 liver expression. Because Acot11 is involved in fatty-acid metabolism and obesity risk, these findings are of importance for the negative impact disturbed sleep has on energy homeostasis in humans. The results link peripheral fatty-acid metabolism with sleep homeostasis and illustrate the power of systems genetics to build hypotheses on mechanistic pathways.
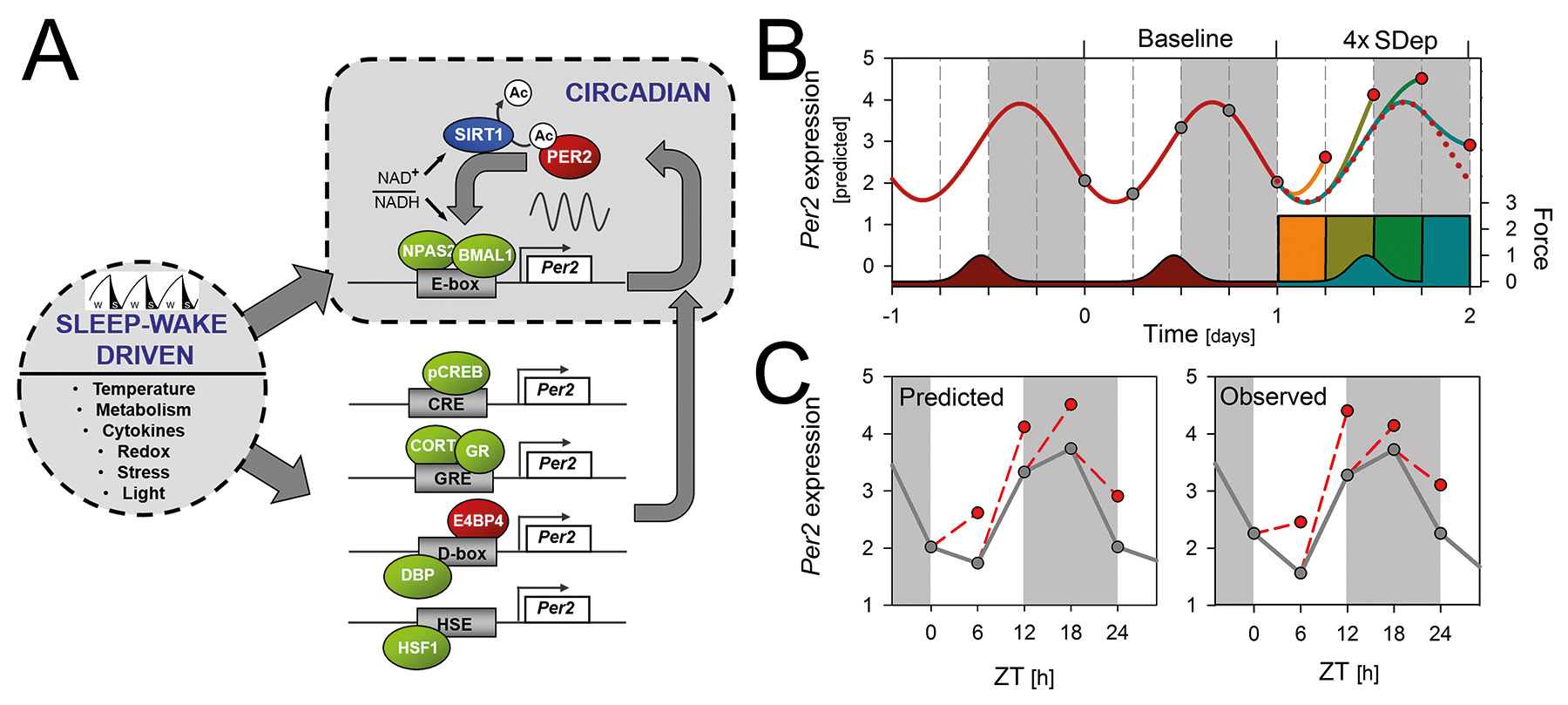
Fig 1. Circadian and sleep–wake driven aspects of Per2 expression.
A) Summary of the molecular mechanisms by which the sleep–wake distribution could alter clock gene expression in the forebrain, illustrated for Per2. A negative-feedback loop underlies the circadian rhythm of Per2 with NPAS2::BMAL1 heterodimers activating Per2 transcription through E-box enhancers. In turn, Per2 protein, through association with NPAS2-BMAL1, inhibits its own transcription. Per2 expression is, however, also regulated by a number of sleep–wake dependent factors (e.g., redox changes, metabolism, cytokine levels, and temperature) acting through cAMP- (CRE) and glucocorticoid- (GRE) response elements, heat-shock elements (HSE), and D-box enhancers. These sleep–wake driven Per2 changes impinge on the ongoing circadian oscillation as conceptualized in the model presented in panel B.
B) Mathematical simulation of the time course of Per2 expression in the brain assuming a driven and damped harmonic oscillator with as periodic driving forces the sleep–wake distribution and changes in corticosterone levels. These recurrent forces initiate and set the phase of the circadian expression pattern in baseline (dark-red line). SDep exert a force altering Per2 expression in forebrain depending on the times-of-day SDep was performed (orange, dark-yellow, green, and blue colored bars and lines).
C) The model accurately predicts (left) the observed (right panel) SDep-induced changes in Per2. Gray and red symbols represent Per2 levels at the time points samples were taken during baseline and after SDep, respectively. Light gray areas denote the dark periods. [see Franken, Curr Opin Neurobiol 23:864-72, 2013, Curie et al., Sleep 36:311-23, 2013]
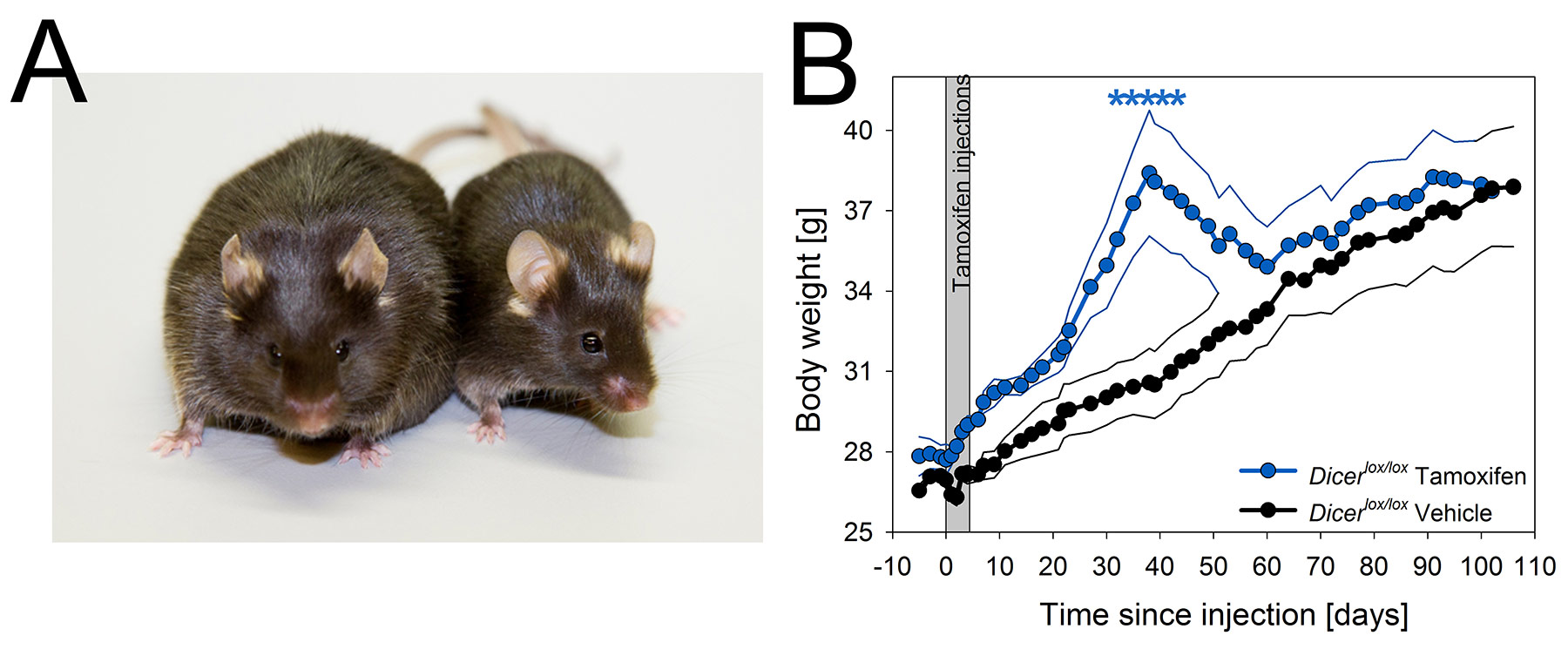
Fig 2. Dicer conditional KO mice show severe but transient obesity.
A) Photograph of two representative Cre+;Dicerlox/lox mice injected either with tamoxifen (cKO, 55.2 g) or vehicle (27.2 g) taken 49 days after injection. Mice are male littermates.
B) Time course of body weight in cKO and vehicle injected control mice. Gray panel marks the 5 days of tamoxifen treatment. cKO mice increased their body weight until reaching a maximum at day 38. Values reverted to control levels within 3 weeks. Mean values for cKO and control mice are represented in blue and black, respectively. Thinner lines span ± 1 SEM. Stars mark significant differences (p<0.05). [see Mang et al., PLoS ONE 10:e0116760, 2015]
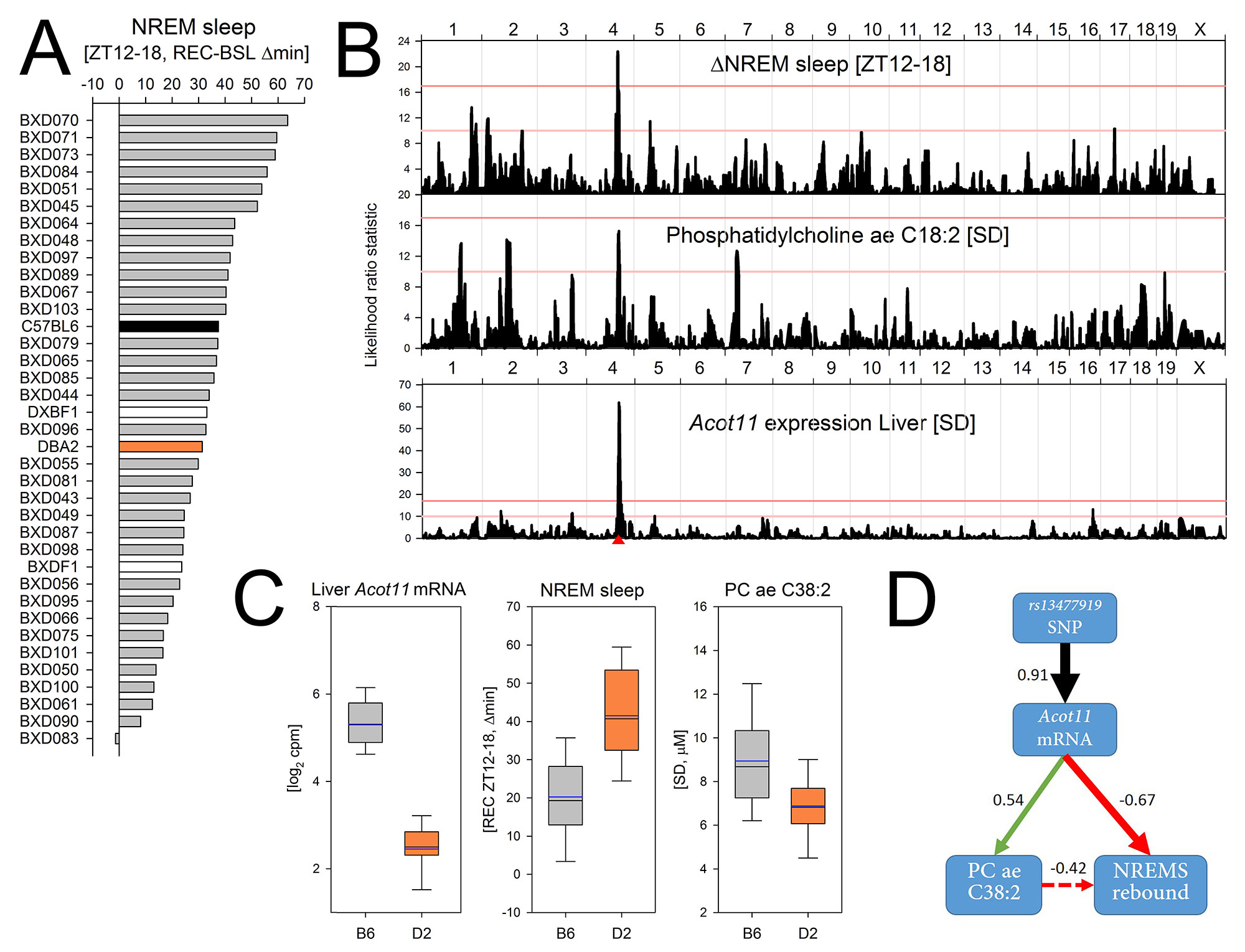
Fig 3. Systems genetics approach to identify the molecular pathways underlying the recovery of sleep.
A) Mice compensate for sleep time lost during SDep by sleeping more during recovery (REC) than during baseline (BSL), especially during the first 6h of the dark period (ZT12-18). The NREM sleep gained during this period strongly varied among the BXD lines from +63 min to -2 min.
B) For this trait a significant QTL was found on Chr 4 (upper panel). Unbiased priority scoring identified Acot11 as top candidate within the Chr 4 QTL. Acot11 encodes an acyl-CoA thioesterase involved in fatty-acid metabolism and obesity risk. Acot11 expression is down-regulated with SDep in liver but not in cortex (not shown) and its liver expression after SDep was under a highly significant cis-eQTL (lower panel; red triangle marks Acot11’s position). A suggestive QTL for plasma levels of the fatty acid Phosphatidylcholine (PC)aeC38:2 after SDep mapped to the same QTL (middle panel).
C) The B6 allele (gray) at the most significant SNP within the QTL (rs13477919) increased Acot11 expression 4.5-fold and PCaeC38:2 levels 1.3-fold, and reduced the NREMS rebound 2.1-fold, compared to the D2 allele (orange, boxplots).
D) Diagram connecting genotype, mRNA, metabolite, and phenotype (with correlation coefficients).
[Diessler, Jan, et al., submitted, 2017]
GROUP MEMBERS
Group Leader
Paul Franken
paul.franken@unil.ch
Postdoctoral Fellows
Charlotte Hor
Jeffrey Hubbard
PhD students
Shanaz Diessler
Marieke Hoekstra
Maxime Jan
Konstantinos Kompotis
Géraldine Mang
Master Student
Mathias Peuvrier
Technicians
Yann Emmenegger
Sonia Jimenez
Adninistrative assistants
Annick Crevoisier
Iris Marouani
iris.marouani@unil.ch