
Short Biography
Christophe obtained his Masters in Biology (2003) and PhD in Computer Science (2009) at ETH Zurich. He stayed at ETH Zurich to become teaching faculty (2010) and senior research associate (2011). In autumn 2011, he moved to EMBL-European Bioinformatics Institute in Hinxton (Cambridge, UK) on a fellowship of the Swiss National Science Foundation. In 2013, Christophe joined University College London (UCL) as a Lecturer — on a joint appointment between Biosciences and Computer Science — and was promoted to Reader in 2015. In late 2015, he joined the CIG as a Swiss National Science Foundation professor. He is also affiliated to the Dept. of Ecology and Evolution at UNIL, to the Swiss Institute of Bioinformatics, and retains an appointment at UCL where part of his lab remains active. Christophe’s contributions were recognized by the SIB Young Bioinformatician Award (2012), a Google Faculty Award (2015), and the EMBO Young Investigator program (2016).

CHRISTOPHE DESSIMOZ – RESEARCH REPORT
Computational Evolutionary Biology and Genomics
At the interface between biology and computer science, our laboratory seeks to better understand evolutionary and functional relationships between genes, genomes and species. A few key underlying questions are:
- How can we extrapolate to the rest of life, and in the best way possible, our current knowledge in molecular biology while concentrating on just a handful of model organisms?
- Conversely, how can we exploit the wealth and diversity of life to get a better grasp on specific organisms or systems of interest?
- Can we summarize meaningfully the evolutionary history of species by arranging them into a small number of tree topologies that capture both vertical inheritance and the most important events of non-vertical inheritance?
Our activities are divided between bioinformatics methods and resource development, and their application – typically with experimentalists.
Recent highlights
Because many genes are highly conserved in sequence and function across different species — in some cases despite billions of years of intervening evolution — knowledge painstakingly gleaned through experiments can often be propagated across evolutionarily related genes. These corresponding genes — called “orthologs” — can however be challenging to infer, with different methods leading to different results. We lead the Quest for Orthologs consortium benchmarking working group in establishing minimum standards in orthology benchmarking and in reporting the outcome of a community experiment including 14 leading orthology methods (Altenhoff et al., Nature Methods 2016). For these, a battery of 20 tests was carried out on a common set of 66 genomes crossing all kingdoms of life. Such analyses can now be easily performed using the orthology benchmark website, which will guide the development of improved orthology inference methods in the future. To learn more about this project, refer to this blog post.
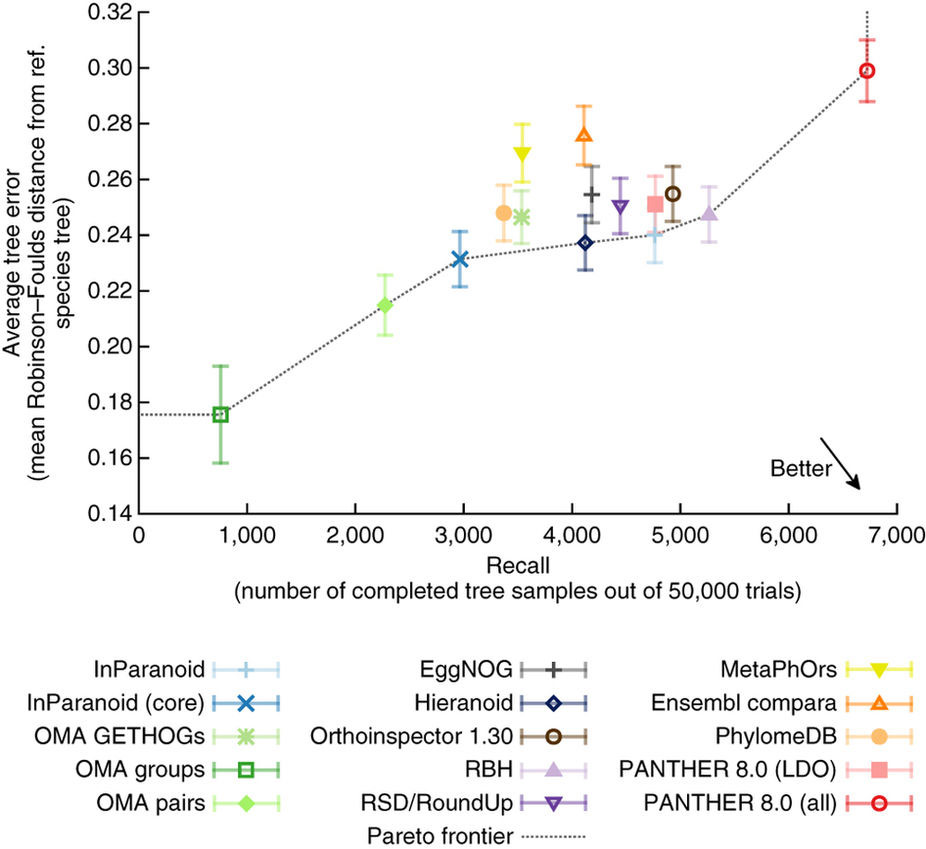
Fig 1. Results on one of the benchmark. A trade-off between precision (measured in terms of tree error in the y-axis) and recall (measured in terms of completed tree samples in the x-axis) can be observed. OMA, developed in our lab, is particularly stringent.
In a collaboration with colleagues from University College London, we reported that membrane proteins are shared by dramatically fewer species than water-soluble proteins across the entire tree of life, on the basis of a comprehensive computational analysis of protein orthologs (Sojo et al., Mol Biol Evol 2016). This pattern holds true within each of the three domains of bacteria, archaea and eukaryotes (Fig 2). These striking differences in conservation of membrane proteins versus water-soluble proteins have implications for evolution and medicine. For example, over half of all known drug targets are membrane proteins, so our findings could help explain why the progression of new drugs from animal models into human trials is often unsuccessful. Our results are also of practical importance in phylogenetics: if membrane proteins are less than half as likely to be conserved widely across the tree of life, homology searches will often be confounded, as will molecular clocks.
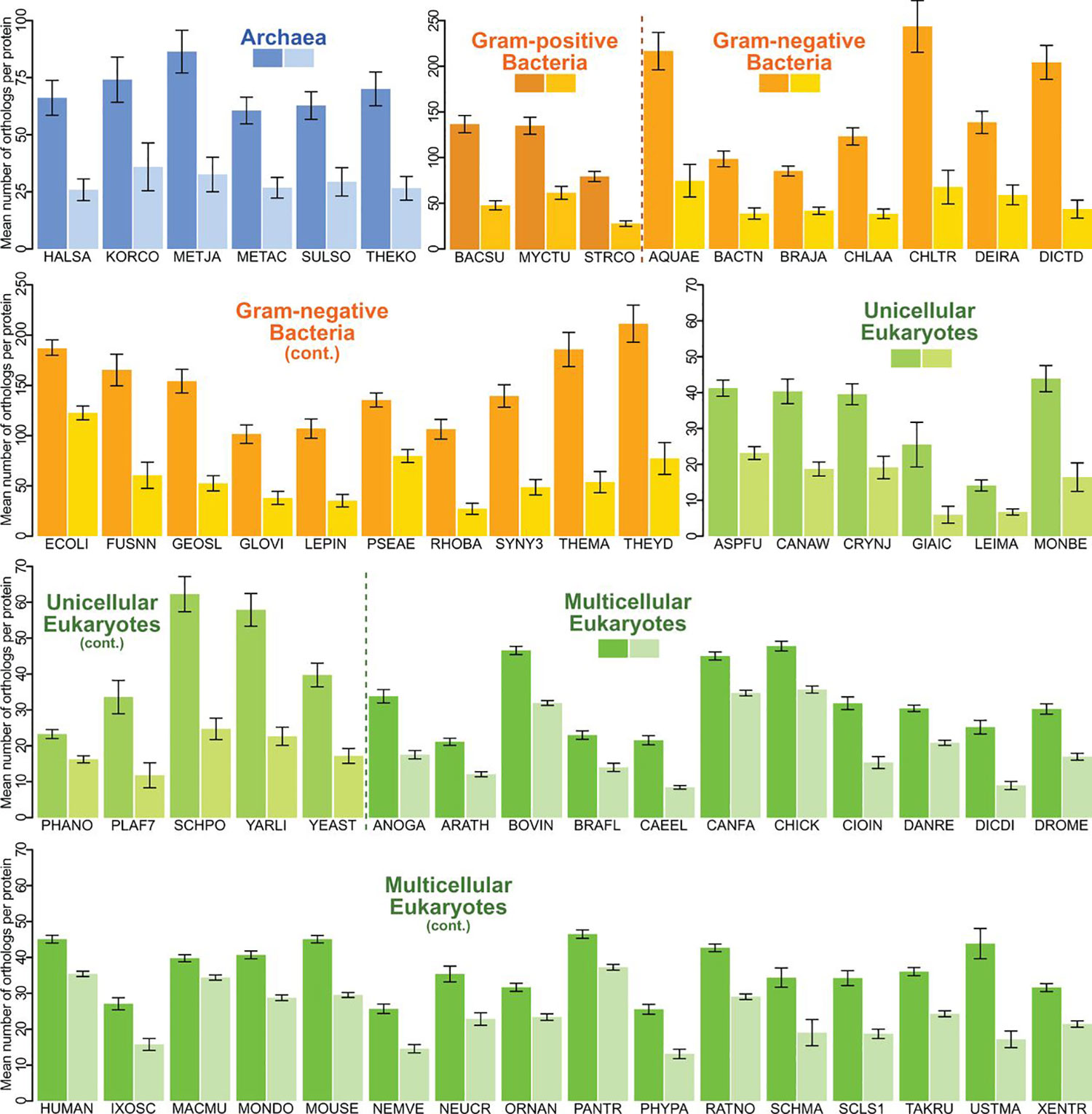
Fig 2. Membrane proteins (light shade) have fewer orthologs than cytosolic counterparts (dark shade) in all three domains of life. Source.
Unlike humans, who have two copies of each chromosome, many plant genomes have more than two corresponding chromosomes. These are called “polyploids”. In a high-profile review paper (Glover et al., Trends Plant Sci 2016), we established a clear and tractable definition for the concept of « homoeology”, which refers to pairs of related genes within a same genome that arise through hybridization (“allopolyploidization”) of the genome. To learn more about this project, refer to this blog post.
We published The Gene Ontology Handbook. The book provides a practical and self-contained overview of the Gene Ontology— the leading project to organize biological knowledge on genes and their products across genomic resources, and thus an essential resource in any bioinformatician’s toolbox. The electronic version is freely accessible (open access).
Tools and resources developed in the lab
One important mission of the lab is to develop tools and resources to facilitate and accelerate biological discovery. These are some of our most widely used tools:
- The OMA database (Orthologous Matrix) establishes evolutionary relationships among genes from about 2000 species, and provides a website to explore these relationships.
- OMA Standalone is the software version, useful to run the OMA algorithm on your own data.
- ALF is a powerful yet easy-to-use gene/genome sequence simulation tool (available both as downloadable version and web interface).
- SWPS3 is a highly optimised Smith-Waterman implementation for x86 and IBM Cell B.E. architectures.
- The Orthology Benchmark Service (mentioned in the highlights above) is a web-based server to assess the quality of ortholog predictions.
- Phylo.io is a web-based phylogenetic tree visualisation tool with powerful side-by-side comparison features.
GROUP MEMBERS
Group leader
Christophe Dessimoz
christophe.dessimoz@unil.ch
Senior Scientist
Adrian Altenhoff
Postdoctoral fellows
Leonardo de Olivera Martins
David Dylus
Natasha Glover
Nives Škunca
PhD Students
Kevin Gori
Jeremy Levy
Ivana Piližota
Clément-Marie Train
Alex Warwick Vesztrocy
Karina Zile
Visiting Scientists
Raphaëlle Luisier
First-Step Student
Victor Rossier
Summer Students
Jan Koch
Abraham Olaoye
Oscar Robinson
Administrative Assistants
Anne Cuendet
Julie Papet
julie.papet@unil.ch
Suzanne Soto
suzanne.soto@unil.ch